Gaussian计算案例
结构优化
GaussView6.0:
输入文件格式:nprocshared–核;mem–内存;%chk–检查点文件;#p–计算类型;opt–优化;
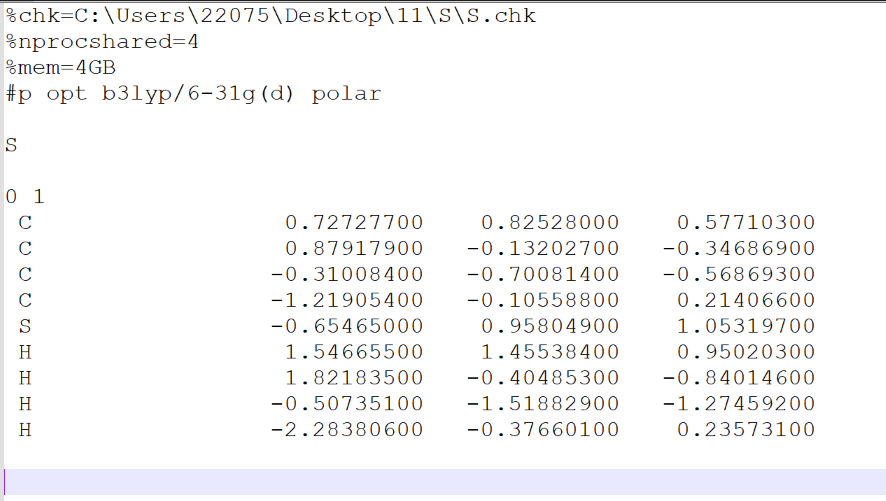
输入文件编辑完成后,用Gaussian16打开进行计算。
Gaussian16计算完成后,通过Utilities–FormChk,将.chk文件转换为.fch文件格式
.chk文件格式无法通过记事本形式打开,会出现乱码;.fch文件格式可以通过记事本形式打开##
Gaussian16:
打开.chk,最好是.fch文件,即可观察到优化后的分子模型。
.chk是过程文件;.out是最终输出文件
.out文件:
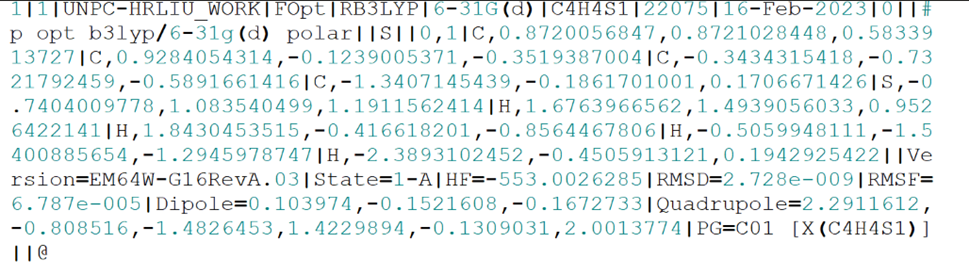
通过文本编辑器将.out输出文件打开,里面包含所需的各种数据信息。
cycle为循环次数,error收敛至D^-8^数量级时将停止计算,返回进行各项条件验证。

对力和位移所需要的条件进行对比,条件符合,循环停止,计算停止,输出最后计算结果;条件不符合,重启循环,循环次数+1,循环完成后再次进行对比。

当所需条件全部满足后,循环停止,计算停止,出现Optimization completed,代表优化过程结束,之后输出数据即为优化后所得各项数据。
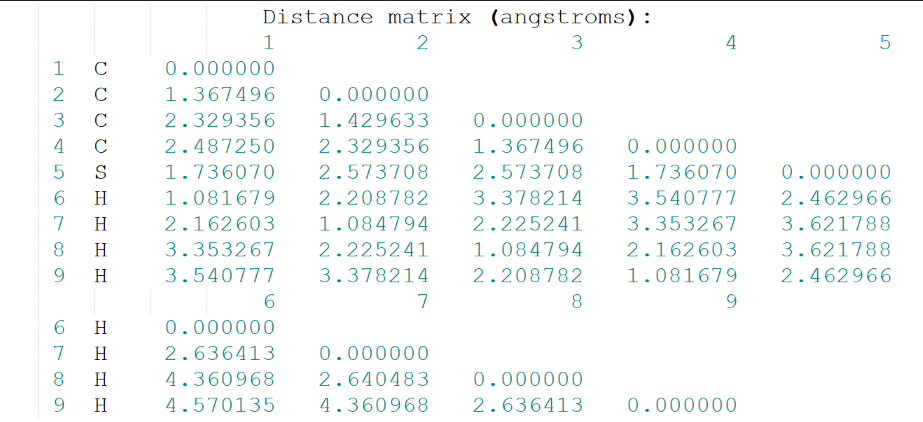
距离矩阵:
轨道对称性:

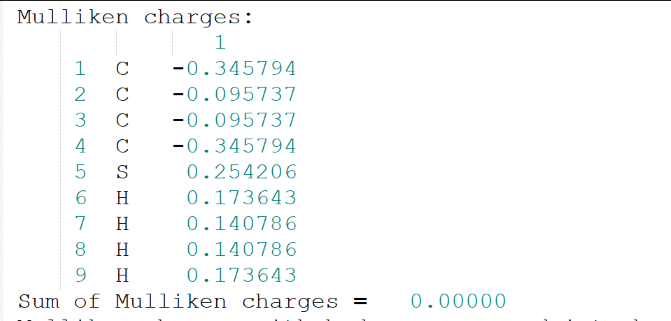
Mulliken电荷:
 偶极矩&多极矩:
偶极矩&多极矩:
 Hartree-Fock能量:
Hartree-Fock能量:
 此项代表计算过程无误正式结束:
此项代表计算过程无误正式结束:
原子单位(a.u,)*27.2114 = 电子伏(eV)##
*gap = |HOMO - LUMO| 能隙分子从基态到激发态所需的最小能量,代表着分子被激发的难易程度##
振动光谱( IR&Raman )
GaussView6.0:
同理,在优化后的.out文件上进行计算文件编辑,得到raram.gjf文件。
输入文件设置如下:
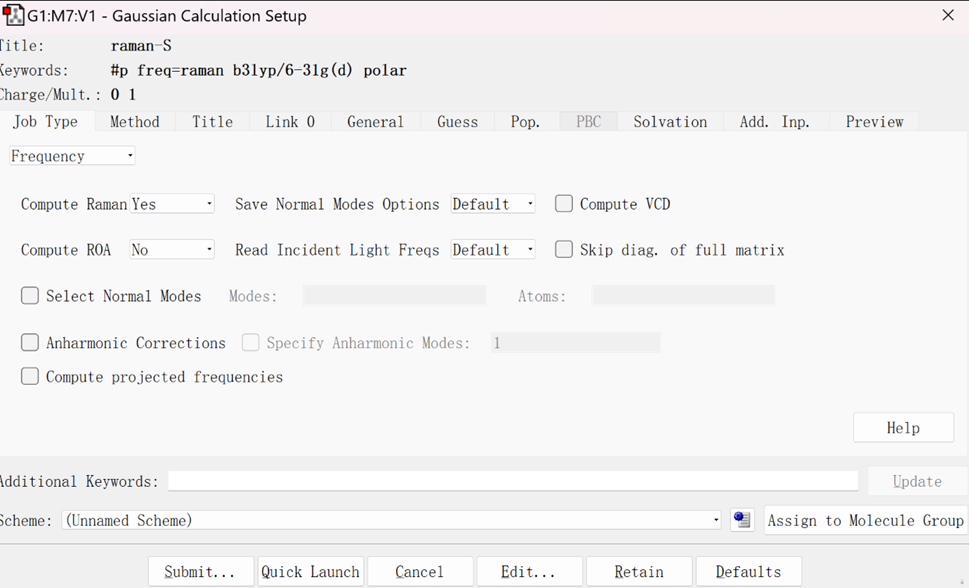
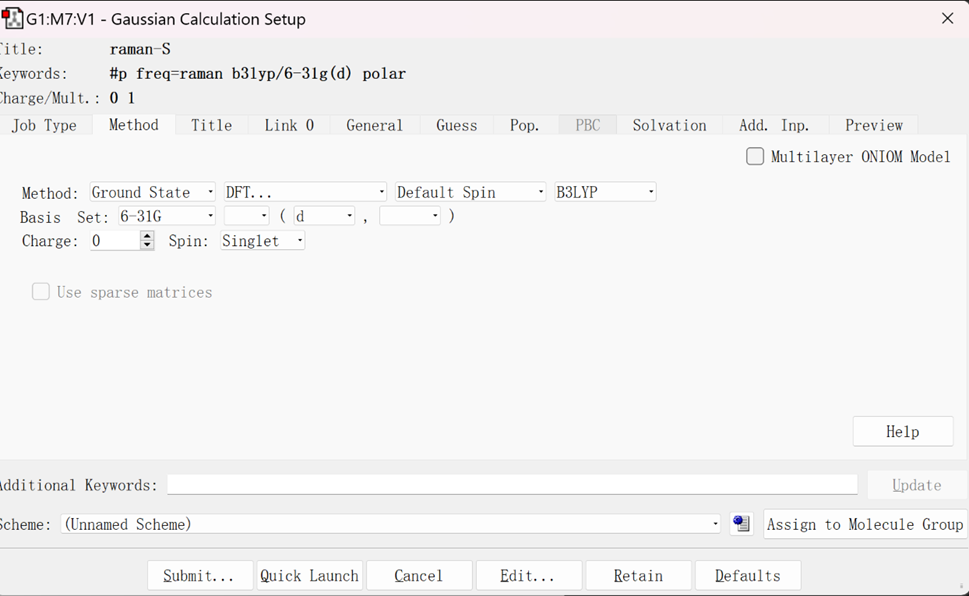
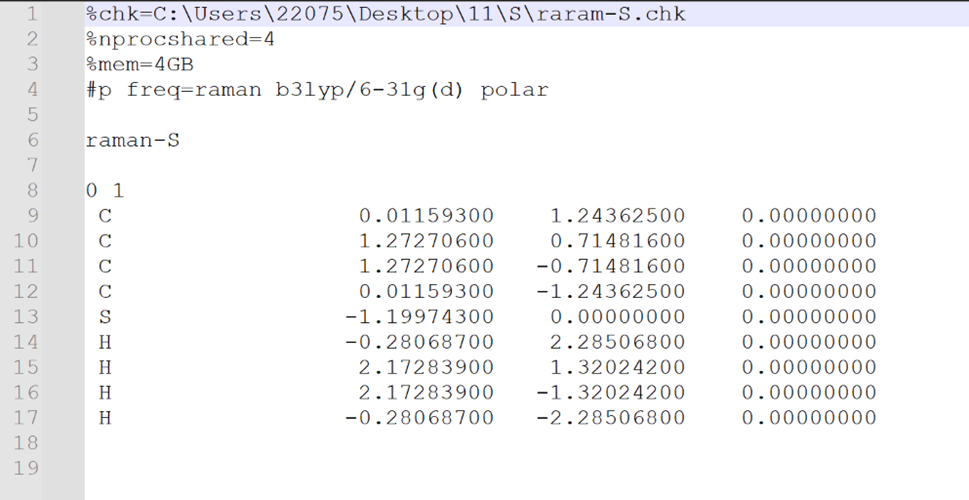
选择Frequency选项进行计算
输入文件编辑完成后,用Gaussian16打开进行计算。
Gaussian16计算完成后,通过Utilities–FormChk,将.chk文件转换为.fch文件格式。
.chk文件格式无法通过记事本形式打开,会出现乱码;.fch文件格式可以通过记事本形式打开
Gaussian16:
打开.out文件,在Results选项中选择Vibartions选项,再选择Spectrum选项,即可得到该分子体系的红外光谱和拉曼活性光谱,如下所示:
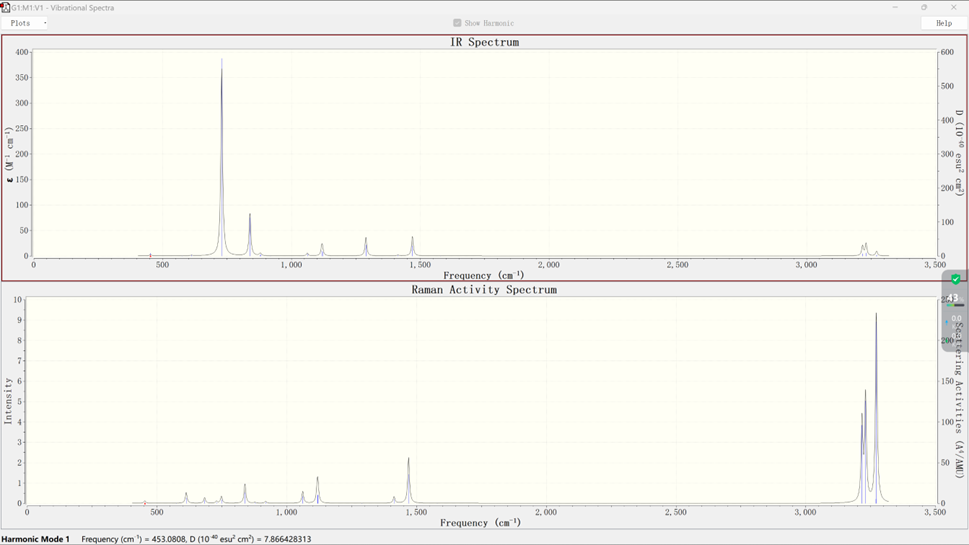
通过光谱寻找感兴趣的模式,在Vibartions选项中可以观察分子振动等各项情况,如下图所示:
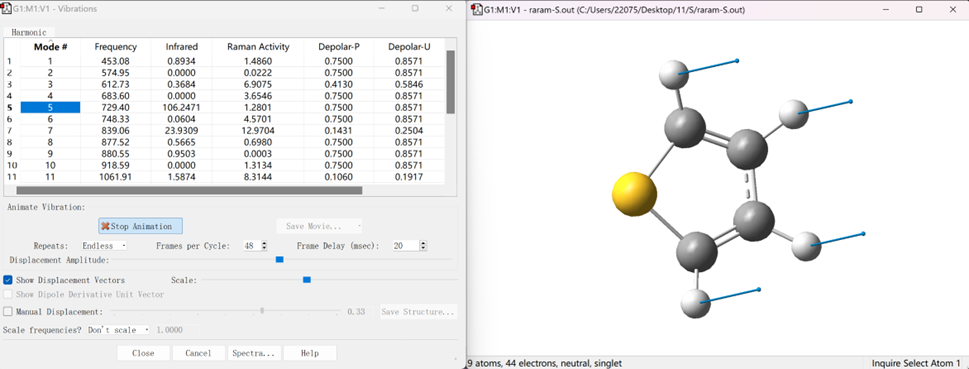
点击鼠标右键,选择Save Data,即可将分子光谱数据保存为文本格式,后续可采用Origin软件进行绘图编辑,保存数据格式如下:
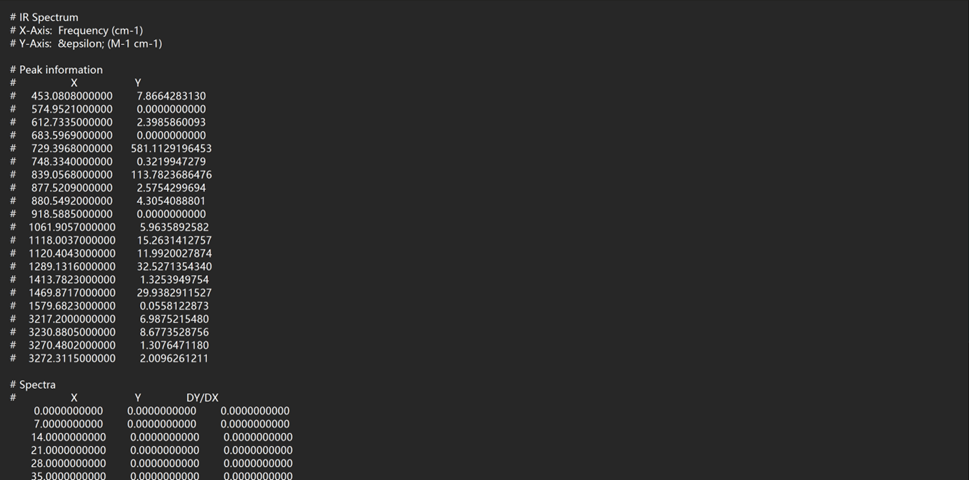
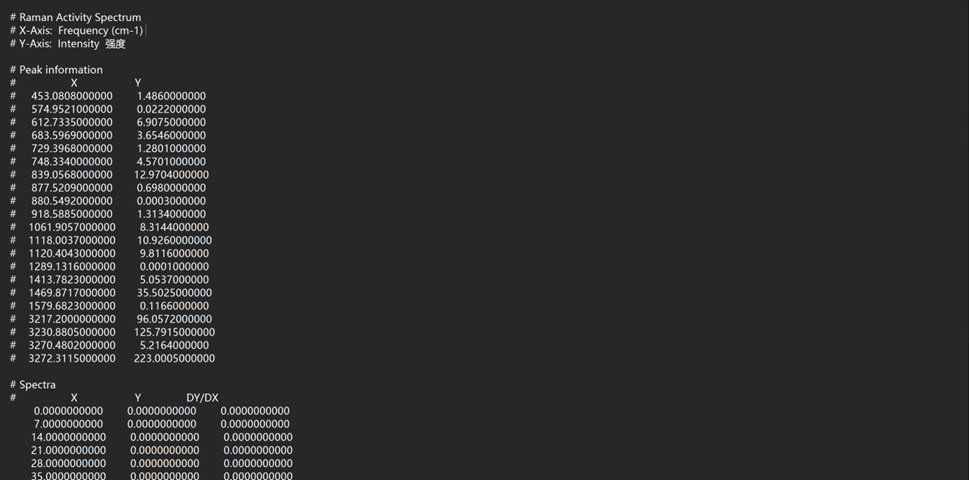
注意:
==我们要对频率一项进行检查,确保没有虚频模式的存在,即频率数值为负数。==
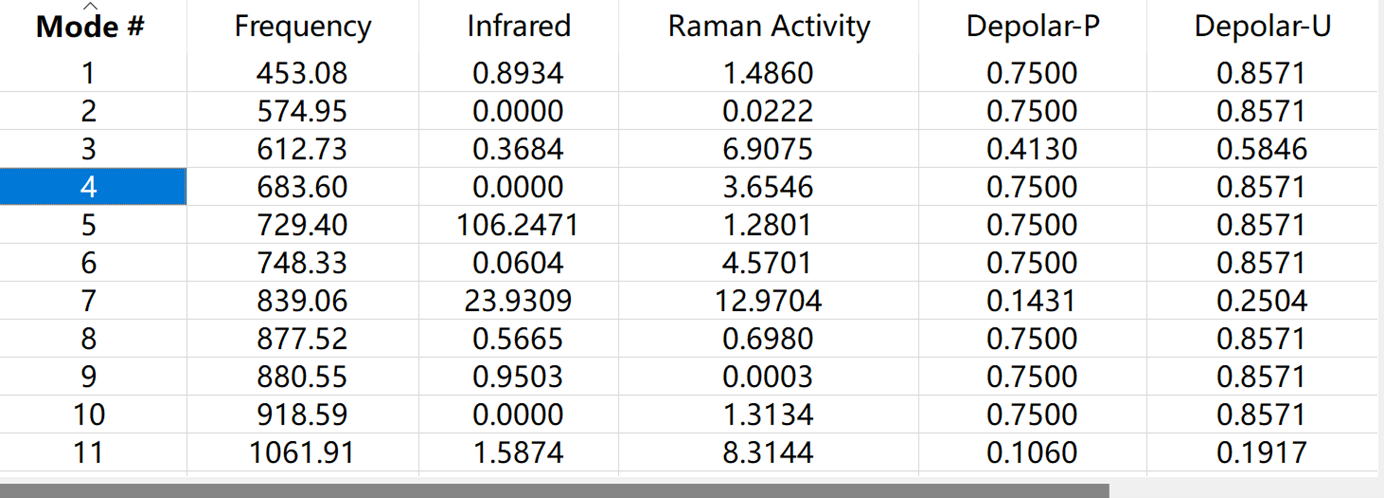
若有虚频模式存在,需重新调整一下分子的几何结构模型,从头开始优化,重复上述过程,直至虚频模式消失。
紫外—可见光光谱( UV-Vis )
进行光谱计算前,要先完成基态能量优化步骤,在所生成的.out文件基础之上进行计算的设计,保存为td.gjf文件,进行UV-Vis计算##
GaussView6.0:
输入文件格式: nprocshared=核数;mem=内存##
Method模块:N=n 激发态数;root=m 感兴趣激发态。
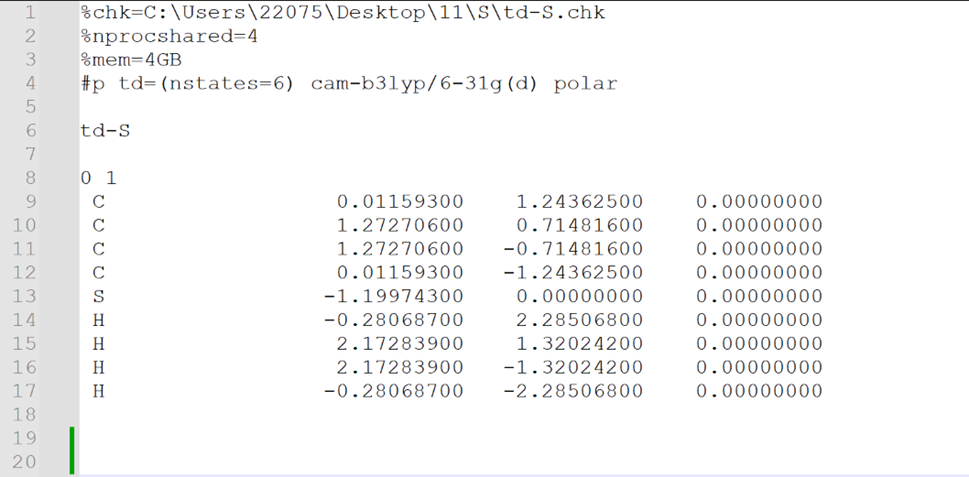
选择Energy选项进行计算
输入文件编辑完成后,用Gaussian16打开进行计算。
Gaussian16计算完成后,通过Utilities–FormChk,将.chk文件转换为.fch文件格式。
.chk文件格式无法通过记事本形式打开,会出现乱码;.fch文件格式可以通过记事本形式打开##
Gaussian16:
打开.out文件,在Results选项中选择UV-Vis选项,即可得到该分子体系的紫外-可见光光谱,如下所示:
上图为紫外-可见光光谱;下图为电子圆二色性谱
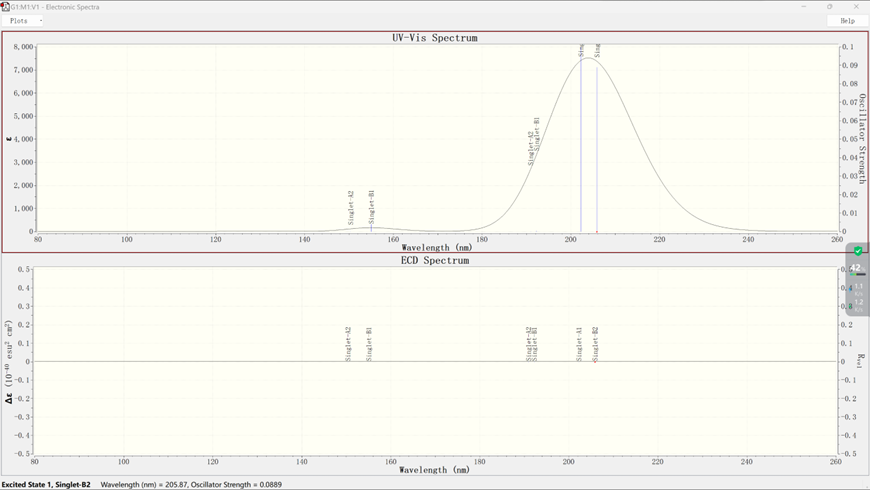
点击鼠标右键,选择Save Data,即可将分子光谱数据保存为文本格式,后续可采用Origin软件进行绘图编辑,保存数据格式如下:
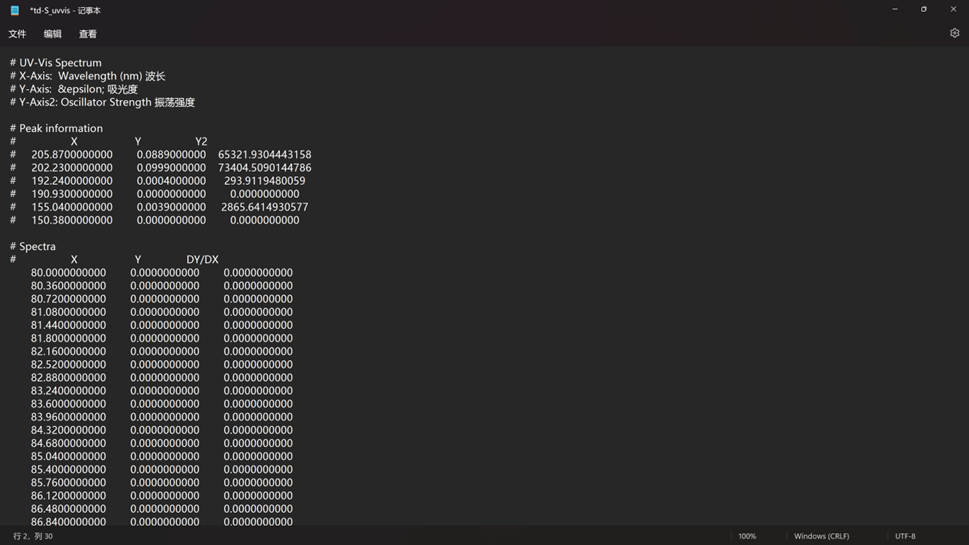
.out文件:
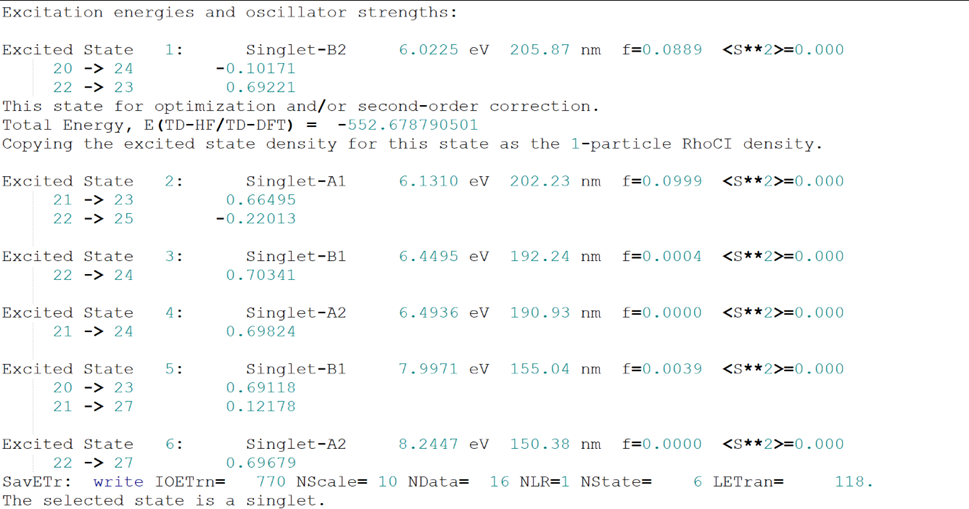
对.out文件进行分析,我们可以得到很有有用的信息。
关于分子体系激发态的一些信息如下图所示: